







(Este artículo se publica con autorización del Policlínico Regional “J.D. Perón” Villa Mercedes – San Luis y de los padres del paciente.)

Jefe De Servicio Oftalmología
Policlínico Regional J. D. Perón Villa
Mercedes San Luis, Argentina
Resumen:
Los teratomas son tumores compuestos de tejidos derivados de las tres capas germinales que pueden localizarse en las gónadas o extragonadales. Los de localización orbitaria son infrecuentes y generalmente se presentan en recién nacidos sanos produciendo proptosis.
Se presenta un caso de un recién nacido varón, sano con una gran proptosis unilateral derecha compatible con teratoma orbitario congénito que invadía las estructuras perioculares y mínimamente intraoculares y que se extendía hacia el cono muscular y espacio orbitario (retro y periocular) con desviación de la línea media orbitaria. Se describen las características clínicas, los hallazgos radiológicos, el abordaje quirúrgico, la descripción histopatoló- gica y el tratamiento adyuvante con quimioterapia recibido por el paciente.
Introducción
Los tumores de células germinales son poco frecuentes en la edad pediátrica; tienen una tasa de incidencia de 2,4 casos/millón/año y representan del 2% al 3% de los cánceres en niños y adolescentes menores de 15 años. Son un grupo de tumores altamente heterogéneos derivados de las células germinales primitivas. Histológicamente pueden ser clasificados como neoplasias benignas o estar constituidos por elementos malignos. Se encuentran en una variedad de sitios pero de acuerdo con su localización se clasifican en gonadales y extra gonadales, estos últimos ubicados en la línea media, como el área sacrocoxígea, retroperitoneo, mediastino, cabeza y cuello y región pineal.
El teratoma es el tumor neonatal más común (aproximadamente 25% de todos los tumores que se presentan en el neonato) y es el tumor de células germinales más frecuente en los niños. Son más frecuentes en el género femenino con una razón femenino/masculino de 3:1. Se localizan mayoritariamente en la región sacro coxígea. Traper D y Lack EE (1983) informaron en la serie de 128 teratomas perinatales que los teratomas sacrocoxígeos representaron el 79,7%, los teratomas de cuello el 4,7%, en la cara 3,1% y los de órbita 1,6%.
La presentación clínica de los teratomas de cabeza y cuello varía de acuerdo al sitio anatómico donde se localice y pueden ser diagnosticados en el periodo prenatal, mediante visión ecográfica, o posnatal. Los orbitarios se presentan al nacimiento con proptosis unilateral en neonatos por lo demás sanos de término o prematuros; se caracterizan por un rápido crecimiento y ocasionalmente el tumor se extiende hasta la cavidad intracraneana.
Casi todos los tumores de células germinales encontrados en el feto y en el neonato son histológicamente benignos y se diagnostican como teratomas maduros o inmaduros. Ocasionalmente en algunos tumores se puede encontrar un foco microscópico de tumor de seno endodérmico y rara vez elementos de carcinoma embrionario asociado ,rara vez, con el teratoma. A pesar que los teratomas de cabeza y cuello son histológicamente benignos constituyen una enfermedad potencialmente fatal que amenaza la vida del neonato por su crecimiento rápido y por la posibilidad de malignizarse con la edad.
El tratamiento de los teratomas orbitarios es esencialmente quirúrgico y en lo posible realizar un salvamento ocular que permita el desarrollo de la órbita y en algunos casos, preservar la visión. Presentamos un caso de un recién nacido con un teratoma congénito con componentes inmaduros a quien se le realizó excentración de la órbita y quimioterapia adyuvante.
Caso clínico
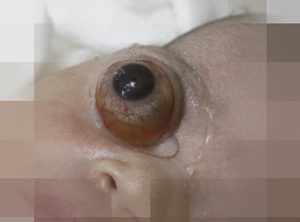 Describimos el caso de un recién nacido masculino de 38 semanas de gestación, controles prenatales normales, informes de ecografías obstétricas sin anomalías y serologías de la madre para sífilis, virus de inmunodeficiencia humana, hepatitis B y C negativas, no se realizó por su condición económica ecografías 4d o 5d . Parto vaginal sin complicaciones; el recién nacido de término con peso al nacer de 3.500 gramos y talla 53 cm.
Describimos el caso de un recién nacido masculino de 38 semanas de gestación, controles prenatales normales, informes de ecografías obstétricas sin anomalías y serologías de la madre para sífilis, virus de inmunodeficiencia humana, hepatitis B y C negativas, no se realizó por su condición económica ecografías 4d o 5d . Parto vaginal sin complicaciones; el recién nacido de término con peso al nacer de 3.500 gramos y talla 53 cm.
Posteriormente al nacimiento se observa gran proptosis de su ojo derecho lo que conlleva a realizar una interconsulta urgente con el servicio de oftalmología.
Al examen presenta proptosis congénita derecha, con imposibilidad de cierre palpebral, sin reacción a la luz, con oftalmoplejia, presencia de buftalmos, quemosis conjuntival, hiperemia.
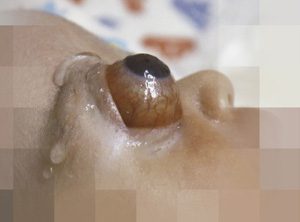 Globo ocular sobresale aproximadamente unos 12 mm, sobrepasando hasta el área frontal y descendiendo por debajo del maxilar produciendo luxación casi completa del ojo con gran compromiso orbitario. Además, desviación de la línea media facial hacia la izquierda (Ver figura 1 frente y figura 2 perfil).
Globo ocular sobresale aproximadamente unos 12 mm, sobrepasando hasta el área frontal y descendiendo por debajo del maxilar produciendo luxación casi completa del ojo con gran compromiso orbitario. Además, desviación de la línea media facial hacia la izquierda (Ver figura 1 frente y figura 2 perfil).
Se le practicó tomografía axial computada y resonancia nuclear magnética cerebral y de órbitas en la que se observó una masa de señal heterogénea de 60 x 60 x 75 mm que reemplazaba el contenido orbitario derecho, con destrucción parcial de las paredes orbitarias, crecimiento exofítico y múltiples áreas quísticas con invasión intracraneal por extensión a través del vértice orbitario hacia la fosa craneal media. Los componentes sólidos de la lesión realzaban de forma heterogénea con el contraste. La órbita izquierda no presentaba alteraciones, en la siguiente imagen por reconstrucción en 3D se observa la falta de unión ósea en pilar externo orbitario derecho y alteración del polo posterior de la órbita debido a la extensión de la masa tumoral. (Ver figuras 3 y 4).
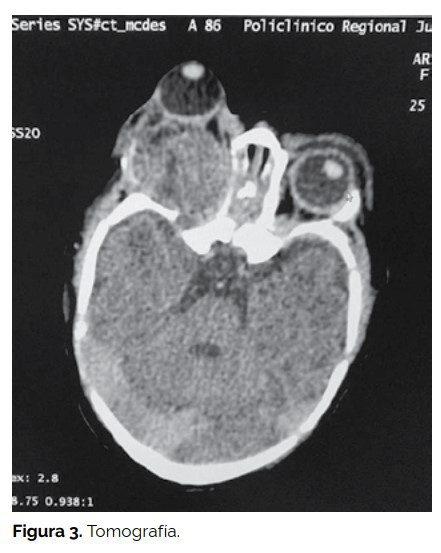
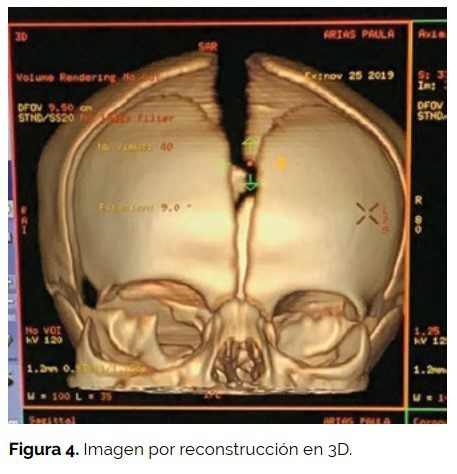
Las radiografías de tórax, ecografía abdominal y ecocardiograma fueron normales.
Los marcadores tumorales pre quirúrgicos: subunidad beta de la hormona gonadotrofina coriónica humana (þ- HCG) fue de 0,935 mUI/l y la alfafetoproteína sérica de 1515 ui/ml; resultados considerados dentro de los límites normales para la edad del paciente.
A los 21 días de vida el paciente fue intervenido por un equipo multidisciplinario integrado por oftalmólogo, neurocirujano y cirujano maxilofacial; encontrándose la presencia de una masa en la órbita derecha de aproximadamente 10 cm de diámetro, ulcerada, eritematosa, de bordes irregulares y encapsulada que producía deformidad de las estructuras óseas perilesionales.
Se realizó exenteración de la órbita derecha con exceresis completa de la tumoración ubicada en la fosa craneal media y en maxilar superior.
En el servicio de patología se recibió la pieza quirúrgica producto de exenteración de la órbita derecha que incluía globo ocular y tejidos blandos vecinos con ampliación de bordes.
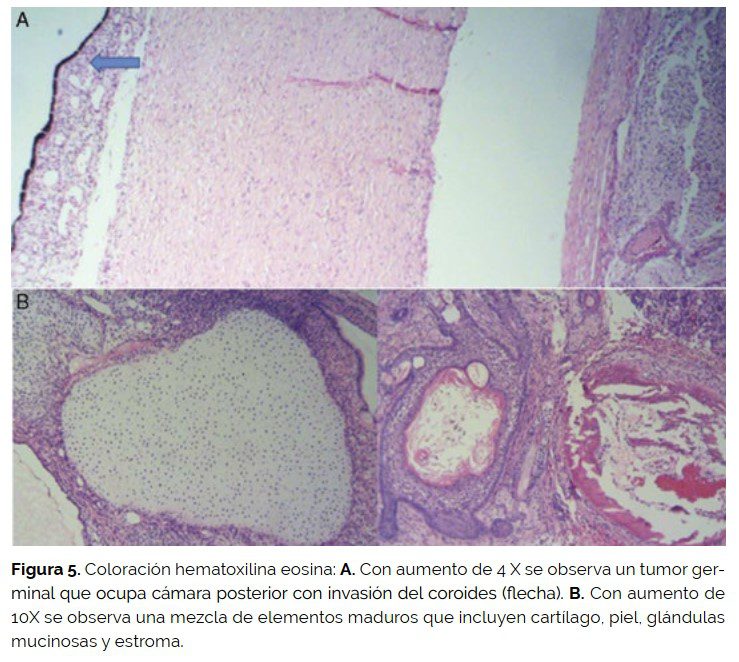
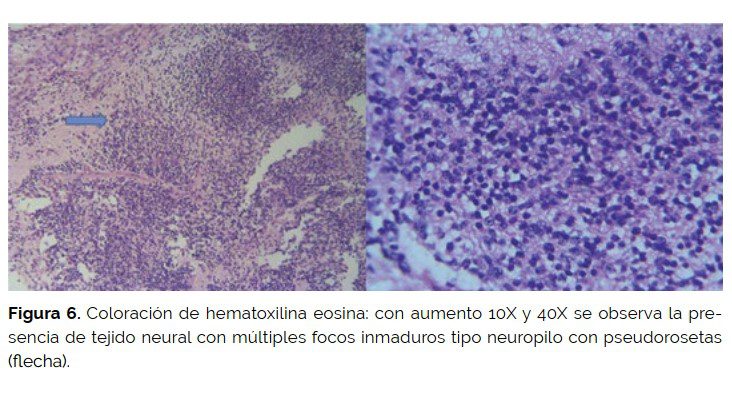
El examen de los cortes microscópicos mostró globo ocular ocupado por un tumor germinal mixto que invadía la cámara posterior y constituida por elementos maduros representados por cartílago, hueso, glándulas mucosas, estroma y piel. Además, se identificó un componente inmaduro formado por tejido nervioso (neuropilo) con formación de pseudorosetas y positividad para CD99 y NSE. En algún corte se observó componente somático de rabdomisarcoma. El tumor tenía invasión de coroides escasa, extraescleral extendiéndose a tejidos blandos vecinos; el borde de sección estaba comprometido por tumor.
El tumor fue clasificado como teratoma inmaduro de órbita grado III con componente somático de rabdomiosarcoma con resección incompleta por bordes quirúrgicos microscópicamente comprometidos (E-III de riesgo intermedio).
Se lo remite a hemato-oncología para tratamiento.
Se le administran dos líneas de tratamiento quimioterapico. -vincristina –actinomicina / Carboplastino – etoposido.
Ante una nueva valoración se observa progresión local del tumor y se le administra Cisplatino/etoposido/ifosfamida (VIP) por 5 días. Continua con progresión local y se decide terapia radiante de órbita y macizo facial.
Al paso de 30 días se controla nuevamente y continúa con progresión tumoral por lo que se administra quimioterapia metronomica con ectoposido por vía oral en ciclos.
Se lo valora nuevamente después de 20 días y se observa progresión facial hacia mejilla y región periauricular.
Ante la evolución del caso se decide informar a los padres para solo continuar con quimioterapia metronomica con ectoposido vía oral explicando efecto adverso y solo con intensión paliativa dada la mala respuesta a los tratamientos instaurados.
Bibliografía
1. Olson TA, Schneider DT, Perlman EJ. Germ cell tumors. En: Pizzo PA, Poplack DC, editores. Principles and Practices of Pediatric Oncology. 6 ed. Philadelphia: Lippinccott Williams and Wilkins; 2011. p. 1045—67.
2. Schneider DT, Olson TA. Germ Cell Tumors of the head and neck. En: Schneider DT, Brecht IB, Olson TA, Ferrari A, editores. Rare tumors in children and adolescents. Ed. Springer-Verlag Berlin Heidelberg; 2012. p. 169—73.
3. Tapper D, Lack EE. Teratomas in infancy and childhood: A 54- year experience at the Children’s Hospital Medical Center. Ann of Surg. 1983;198(3):398—409.
4. Brodsky JR, Kanwar VS, Staff ord LM, Reza R, Frazier L. Germ cell tumors/Teratoma. En: Reza Rahbar, Carlos Rodríguez-Galindo, John G, Maera, Edward R, Smith, Antonio R, Perez-Atayde, edi- tores. Pediatric Head and Neck Tumors. Ed. Springer New York Heidelberg Dordrecht London; 2014. p. 153—63.
5. Sesenna E, Ferri A, Thai E, Magri AS. Huge orbital teratoma with intracranial extension: a case report. J Pediatr Surg. 2010;45(5):E27—31.
6. González C, Restrepo CA, Salazar GI, Monsalve P. Congeni- tal orbital teratoma. Informe de caso Colomb Med. 2012;43: 82—5.







{kind=link}