



Dra. Silvia Moguel Ancheita
Cirujano oftalmólogo, estrabólogo.
Máster en neurociencias
Caso clínico
Caso clínico de una niña con ciliopatía, presentación de Síndrome de Joubert.
Femenina de 3 años, recibida en la consulta del departamento de estrabismo, con antecedente de ser obtenida en gesta 3, habiendo 2 óbitos previos. La abuela paterna con puente nasal deprimido y ptosis bilateral. La madre cursó con preeclampsia y la paciente fue obtenida por cesárea, a las 41 semanas de gestación, con peso al nacer de 2620 g., fue atendida por asfixia neonatal, infección por citomegalovirus, y virus de Epstein-Barr, hiperbilirrubinemia, comunicación interauricular, fibrosis hepática congénita, atresia de vías biliares, paladar hendido posterior. Análisis cromosómico 46XX. Potenciales evocados visuales: P100: OD 156, OI: 99.8
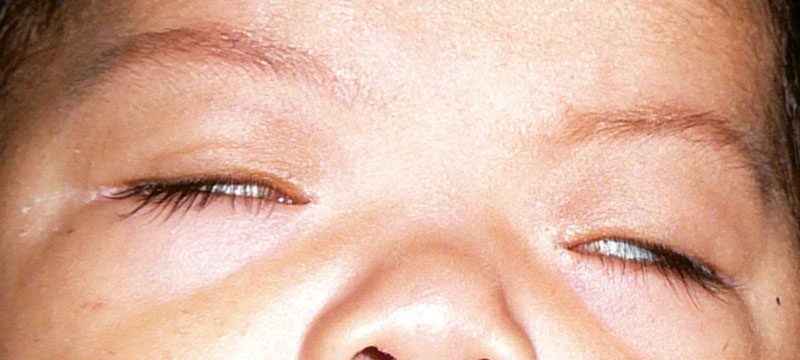
En la exploración se observó síndrome dismórfico, agenesia de huesos nasales, hipotonía facial y generalizada, hipertelorismo, cejas arqueadas, frente ancha, ptosis palpebral congénita. Emplea el músculo frontal para elevar los párpados y abrir los ojos y responde a la sonrisa.
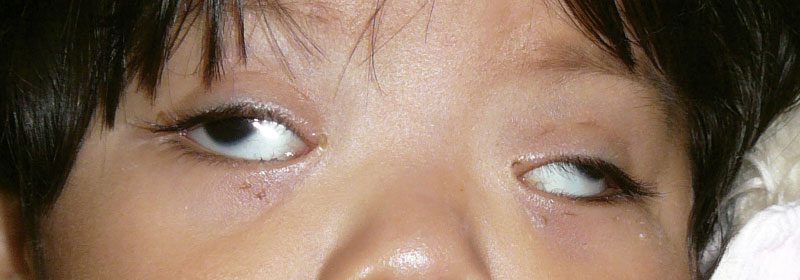
En la exploración oftalmológica se aprecia la ptosis palpebral bilateral, buena atención a la luz con ambos ojos, pupilas midriáticas arrefléxicas, preferencia de fijación ojo derecho, exotropia de gran magnitud de 70 dioptrías prismáticas. Durante los movimientos oculares se observa defecto en ambos ojos con limitación y retracción durante los intentos de aducción y abducción, no hay elevación. Refracción bajo cicloplejia con ciclopentolato: +3.50 esf AO. Electroretinograma con ondas de baja amplitud e intensidad. Retardo en la conducción de VIII par craneal de predominancia izquierda.Ver figuras 1,2.
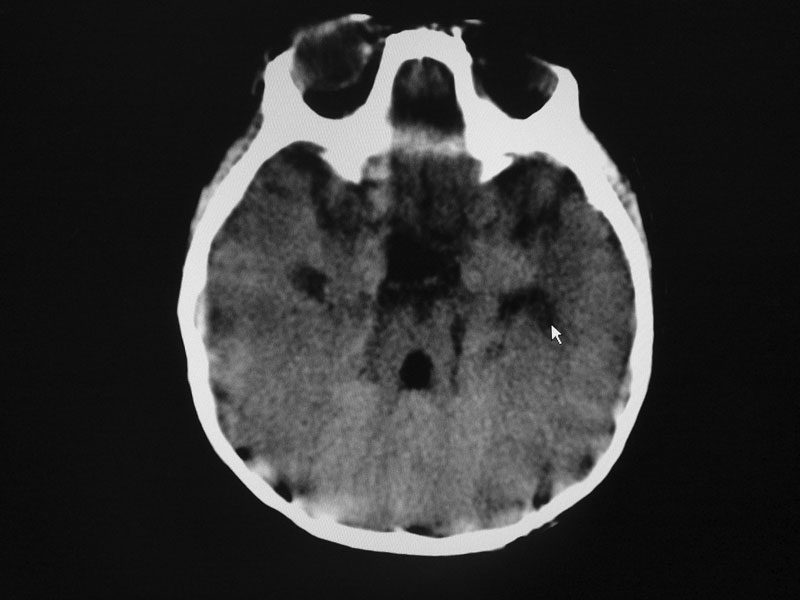
Los estudios de resonancia de cráneo demostraron agenesia de vermis cerebeloso. Se diagnosticó síndrome de Joubert, variedad COACH-Gentile, con rasgos de síndrome Oro-facio-digtal. Ver figura 3.
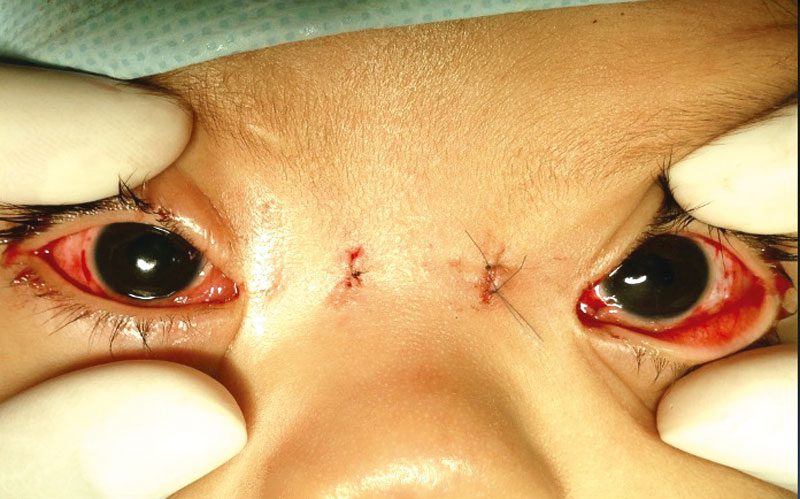
Se realizó tratamiento quirúrgico consistiendo en retroinserción amplia de ambos músculos rectos laterales, y anclaje con sutura no absorbible de tendón de músculos rectos mediales a periostio nasal bajo incisión de piel, bilateral, con lo que se logró mejoría de la posición al frente. Fue vigilada por tres años siguientes hasta su alta del servicio. Ver figura 4.
Discusión
El presente caso se trata de una paciente con afectación multiorgánica caracterizada por instalación muy temprana de las lesiones. El diagnóstico se estableció a través de la imagen correspondiente a la agenesia del vermis cerebeloso, como síndrome de Joubert (SJ). Considerado una neurocrestopatía por ser un daño de los elementos provenientes de la cresta neural, el SJ es también una ciliopatía; que son enfermedades genéticas por afectación del cilio primario, estructura microtubular proteica (axonema) que emerge del cuerpo basal, desde la superficie celular al espacio extracelular, con función de sensor, señalizador y vehículo transmisor de información entre la célula y el espacio extracelular. Es importante en la guía de hormonas, proliferación, migración y supervivencia celular. Es frecuente la disfunción ocasionando neuropatías, retinopatías y nefropatías, con grave riesgo en la supervivencia del embrión. Existen más de 100 ciliopatías y agrupadas pueden considerarse bajo una incidencia de 1:1000. (1,2,3,4)
En el caso del SJ el daño del cilio es en la zona de transición del complejo proteico B9 o complejo tectónico, que evita la rápida difusión de proteínas y receptores no selectivos hacia la zona de transición y promueve el acceso de otras proteínas y receptores específicos.
Fue descrito por Joubert (1969), como una enfermedad multiorgánica compleja, con transmisión autosómica recesiva (subtipo 10 ligado al X recesivo) con 34 subtipos e incidencia de 1:80,000. Prevalece alta frecuencia en canadienses franceses (mutación C5ORF42); en Ashkenazi, incidencia de 1:34,000 (TMEM216), y en Hutteritos (TMEM237) de 1:1150.
Ha sido descrita la afectación en 28 genes, entre ellos AHI1, CPLANE1, CEP290, CC2D2A, CSPP1, INPP5E, KIAA0586, MKS1, NPHP1, RPGRIP1L, TMEM67 TMEM231. Es característica la disgenesia del vermis cerebeloso con la imagen radiológica del signo de la muela. Se asocia a apraxia oculomotora, retraso en el desarrollo psicomotor, ataxia, degeneración retiniana, quistes renales, fibrosis quística y compromiso esquelético. (5,6)
Hunter describió la variedad COACH en 1974, con hipoplasia de vermis Cerebeloso, Oligofrenia, Ataxia congénita, Coloboma y fibrosis Hepática con mutación en MKS3.
La asociación con fibrosis hepática fue descrita por Gentile (1996), variedad 5 de Joubert (tipo 1 o “A”: Joubert puro con ataxia y lesiones neurológicas; tipo 2 ocular, o “B”, predomina amaurosis congénita de Leber; tipo 3 con defectos renales; tipo 4 defectos oculorenales; tipo 5 con defectos hepáticos; tipo 6 orofaciodigitales, antes síndrome de Varadi-Papp). Hay hipodesarrollo de los conductos biliares intrahepáticos durante la formación de la placa bilaminar, definida como “Malformación de la placa ductal” (Brancatti 2009), con remodelación defectuosa de la placa biliar y fibrosis progresiva de los tractos portales. Puede haber hepatoesplenomegalia, hipertensión portal, varices esofágicas y cirrosis. (7,8,9)
La variedad 6 incluye las lesiones oro-facio-digitales, descrito por Mohr en 1941, es un trastorno complejo del desarrollo con 13 formas diferentes, en los tipos I y II se registran fisuras palatinas y anomalías nasales. La mayoría con herencia autosómica recesiva; en el OFD I, el más frecuente y está ligada a X dominante. (10-11)
Los estrabismos asociados a neurocrestopatías y ciliopatías pueden estar relacionados con la formación temprana de los nervios craneales. En este caso, se trata de un estrabismo divergente de gran ángulo con afectación de los III nervios craneales con cambios aberrantes por movimientos retráctiles, hacia arriba, ante el intento del movimiento horizontal. Estos estrabismos pueden resultar ser restrictivos. La falta de función de motoneuronas establece un mal pronóstico a las técnicas clásicas por lo que se decide el anclaje de los rectos mediales hacia elementos de sostén, como el periostio nasal. Este tipo de estrabismos son incluidos en las disgenesias de nervios craneales (CCDD por sus siglas en inglés). (12,13)
Conclusiones
Las anomalías primarias de tejidos provenientes de la cresta neural como las neuocrestopatías y las ciliopatías deben sospecharse en todo niño con lesiones multiorgánicas y fallas de sistemas, malformaciones, hipotonía, movimientos oculares anormales. Es imprescindible el estudio integral incluyendo imágenes que demuestran la formación de los elementos cerebrales; en este caso, fue clave para el mismo ante el hallazgo del signo de la muela, cuya detección prenatal puede determinarse a partir de la semana 17 de gestación.
Bibliografía
- Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, Johnson CA. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel–Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Human Molecular Genetics 2013, 22 (7): 1358–1372 doi:10.1093/hmg/dds546
- Shaheen R, Szymanska K, Basu B, Patel N, Ewida N, Faqeih E, y cols. Characterizing the morbid genome of Ciliopathies. Genome Biology 2016,17:242. DOI 10.1186/s13059-016-1099-5
- Smith CEL, Lake AVR, Johnson CA. Primary Cilia, Ciliogenesis and the Actin Cytoskeleton: A Little Less Resorption, A Little More Actin Please. Front Cell Dev Biol 2020, 17(8):622-822. doi: 10.3389/fcell.2020.622822. PMID: 33392209; PMCID: PMC7773788.
- Ramírez CZ, Zubia DF, González NC. La disfunción del cilio primario y su relación con las ciliopatías. Bases moleculares y celulares. Panorama Cuba y Salud 2017;12(1):45-52
- Dafinger C, Liebau MC, Elsayed SM, Hellenbroich Y, Boltshauser E, Korenke GC, y cols. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J Clin Invest 2011;121(7):2662–2667. doi:10.1172/JCI43639.
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis 2010;5:20.
- Doherty D, Parisi MA, Finn LS, Gunay-Aygun M, Al-Mateen M, Bates D, y cols. Mutations in 3 genes (MKS3, RPGRIP1L, and CC2D2A) cause COACH syndrome/Joubert syndrome with congenital hepatic fibrosis. J Med Genet 2010; 47:8-21.
- Angemi JA, Zuccotti JC. Actualizaciones sobre Sindrome de Joubert. Revista Argentina de Clínica Neuropsiquiátrica 2012, 18(1): 25-37.
- Parisi M, Glass I. Joubert Syndrome. Updated 2017 Jun 29. En: Adam MP, Ardinger HH, Pagon RA, et al., ed. Gene Reviews. Seattle (WA): University of Washington, Seattle; 1993-2022. https://www.ncbi.nlm.nih.gov/books/NBK1325/
- Baraitser M. The orofaciodigital (OFD) syndromes. J Med Genet 1986, 23(2):116-9. doi: 10.1136/jmg.23.2.116. PMID: 3712388; PMCID: PMC1049564.
- Edel T, Zárate-Sanabria AG, Briceño-Balcázar I, Martínez-Lozano JC. Paciente con síndrome oro-facio-digital tipo II. Reporte del caso. Iatreia 2017, 30(1), 86-91[fecha de Consulta 20 de Marzo de 2022]. ISSN: 0121-0793. Disponible en: https://www.redalyc.org/articulo.oa?id=180549475009
- Cintora SM, Sánchez SA, Herrero AM, Carabaño AI, Aguirre PE, Salcedo LE, y cols. Síndrome de Joubert. Rev Pediatr Aten Primaria 2021;23:191-4.
- Basten SG, Giles RH. Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia 2013, 2:6
http://www.ciliajournal.com/content/2/1/6






