![]() Historia clínica
Historia clínica
Paciente femenina de 35 años de edad, acudió a nuestro Instituto por disminución progresiva de la agudeza visual en ambos ojos, mayor en ojo derecho de 6 meses de evolución, sin otros síntomas oftalmológicos, ni sistémicos asociados. Refirió haber sido tratada con acetato de prednisolona tópica y Meticorten® sistémico con dosis de 1 mg por kilo de peso por probable uveítis intermedia dos semanas antes de nuestra valoración. Adicionalmente, la paciente refirió antecedente de hermano finado por falla hepática como complicación de Amiloidosis familiar confirmada por biopsia.
Exploración física
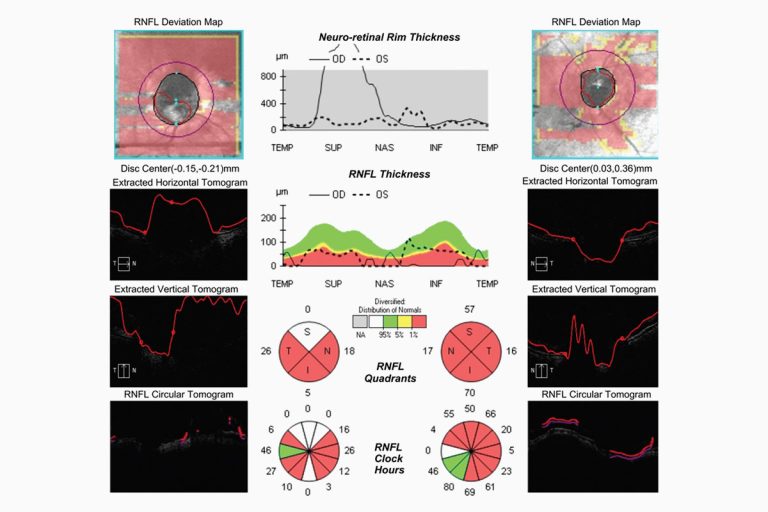
La agudeza visual de la paciente al momento de la valoración fue movimiento de manos a 30 cm en el ojo derecho y 20/70 en el ojo izquierdo. No se encontraron alteraciones en segmento anterior de ningún ojo y la presión intraocular se encontró en 12 mmHg en ambos ojos. A través del examen fondo de ojo, bajo dilatación pupilar, se observó vítreo blanco, homogéneo desde la cara posterior del cristalino que no permitió valorar detalles del polo posterior en el ojo derecho (Foto 1), y en el ojo izquierdo se encontró vítreo blanquecino hacia la periferia con opacidades en forma de bandas blancas en la región cortical, disco óptico redondo con excavación de 4 décimos, emergencia central de vasos, vasculatura central y periférica normal y mácula sin alteraciones (Foto 2).
Estudios complementarios
Los paraclínicos realizados se encontraron dentro de límites normales excepto la velocidad de sedimentación globular que estaba aumentada, radiografía de tórax sin alteraciones, fluorangiografía de ambos ojos sin alteraciones y ultrasonido de ojo derecho en el que se evidenciaron ecos homogéneos de mediana reflectividad en toda la cavidad con retina aplicada (Foto 3).
Conducta
Teniendo en cuenta antecedente familiar de la paciente y la alta sospecha de amiloidosis, se solicitó valoración por médico internista quien sugirió realizar vitrectomía para estudio histopatológico de vítreo obtenido.
Se realizó vitrectomía 27 Gauge convencional bajo anestesia regional y sedación utilizando 3 puertos con equipo Constellation® sin complicaciones, se realizó una valoración exhaustiva de la periferia sin encontrar lesiones o datos de vasculitis y, posteriormente, se hizo intercambio líquido aire (en el siguiente link podrá encontrar el video del manejo quirúrgico de nuestra paciente: https://eyetube.net/video/rpesp/ ).
Se envió muestra obtenida de vítreo durante procedimiento a patología para estudio con inmunotinciones (rojo congo y cristal violeta) que fueron positivos para material amiloide (Foto 4, 5 y 6).
Evolución clínica
Tres meses después, el ojo derecho presentaba una agudeza visual de 20/30 y el ojo izquierdo de 20/70. (ver foto 7. Imagen fondo de ojo en post operatorio). La paciente fue referida para su tratamiento sistémico en forma multidisciplinaria. No se realizó vitrectomía en ojo izquierdo ya que la paciente no aceptó tratamiento en ese momento.
Discusión
Las amiloidosis son un grupo poco frecuente de enfermedades que se caracterizan por el depósito de sustancia amorfa o material amiloide en el espacio extracelular de diversos órganos y tejidos condicionando alteraciones funcionales y estructurales según la localización e intensidad del depósito 1. Alrededor del 75% de los pacientes que la padecen tienen una amiloidosis primaria, el 5% del total presenta una amiloidosis secundaria (asociada a enfermedad), y menos del 5% desarrolla una forma de amiloidosis familiar. El diagnóstico se basa en la sospecha clínica y la demostración de la presencia de la sustancia amiloide en los tejidos. Estos depósitos de amiloide están constituidos por una serie de proteínas fibrilares no relacionadas bioquímicamente entre sí con unas características comunes que incluyen la birrefringencia verde manzana con la luz polarizada al teñirse con rojo congo y la configuración en hoja plegada β-laminar altamente organizada que le confiere propiedades físico-químicas distintivas de las fibrillas amiloides, incluyendo su estabilidad relativa y resistencia a la proteólisis. Los depósitos amiloides también contienen constituyentes no fibrilares como el amiloide P sérico (SAP), glicosaminoglicanos (GAGs), apolipoproteína E, colágeno tipo IV y laminina 2. Se han identificado más de 30 proteínas involucradas con la formación de amiloide en el hombre; sin embargo, el uso reciente de la espectrometría de masas sugiere que muchas más proteínas podrían estar relacionadas 3.
Las manifestaciones clínicas de la enfermedad son inespecificas y variables, y se encuentran determinadas por el órgano o el sistema afectado. de masas sugiere que muchas más proteínas podrían estar relacionadas 3.
![]()
Diferentes estructuras oculares pueden estar implicadas en cualquier subgrupo de amiloidosis sistémica o en casos poco frecuentes presentarse como un proceso aislado. La polineuropatía amiloidótica familiar (FAP) es una amiloidosis autosómica dominante con penetrancia incompleta y expresividad variable. La primera descripción se hizo en Portugal, en 1952, donde se encuentra la mayoría de pacientes con esta enfermedad 4. Esta patología es causada por una mutación en el gen que codifica la transtirretina (TTR), una proteína inestable se deposita en diferentes tejidos y órganos como fibras amiloides y la característica clínica principal es una polineuropatía periférica prominente.
Se han identificado más de 100 mutaciones puntuales de las cuales aproximadamente 80 se asocian con FAP. La variante más frecuentemente implicada tiene una sustitución de metionina por valina en la posición 30 de la molécula de (TTR V30M). Esta proteína es producida, principalmente, por el hígado, sin embargo también es producida por la retina y el plexo coroideo 5. Se ha reportado que las manifestaciones oculares se presentan en aproximadamente 10% de los casos de FAP y la incidencia de opacidades vítreas varía del 5.4% al 35%(6, 7).
El depósito de amiloide en el vítreo es una manifestación que podría considerarse típica de FAP; sin embargo, se han descrito algunos casos en los que el compromiso vítreo es aislado y no hay compromiso sistémico ni antecedentes familiares de enfermedad considerándose una variante extremadamente infrecuente como en el caso de nuestra paciente. Los signos característicos de esta enfermedad incluyen opacidades vítreas que a menudo son bilaterales pero asimétricas, que tienen apariencia de “vidrio molido” o de “telaraña” y la densidad de estas determina en gran medida la severidad de los síntomas visuales que incluyen: glare, visión borrosa, flotadores y disminución de la agudeza visual. Estas opacidades se extienden usualmente desde la corteza vítrea hacia el centro y son a menudo el único signo de afectación ocular; sin embargo, pueden estar asociadas con otros signos como depósitos en el iris, infiltración coroidea y glaucoma amiloi- de debido a depósitos perivasculares de sustancia fibrilar en la conjuntiva y epiesclera que generan un aumento de la presión venosa episcleral, depósitos de amiloide en el borde pupilar y en la malla trabecular. Otros signos importantes de amiloidosis vítrea son los depósitos perivasculares y la pseudopodia lentis (opacidades blancas puntiformes en la cápsula posterior del cristalino). Los vasos retinianos generalmente son normales, aunque en ocasiones pueden haber depósitos perivasculares que aparecen como placas focales generando algún grado de tortuosidad vascular 8.
El tratamiento quirúrgico de esta patología se introdujo en 1950 con intentos fallidos de aspiraciones del cuerpo vítreo. En 1968 Kasner describe la vitrectomía a cielo abierto utilizando esponjas de celulosa y tijeras a través de una incisión corneoscleral de 300º después de la extracción del cristalino como un procedimiento exitoso pero al mismo tiempo complejo con una importante tasa de complicaciones como desprendimiento de retina, glaucoma y queratopatía bullosa. El desarrollo de la técnología ha facilitado el uso de procedimientos menos invasivos y por tanto más seguros para el paciente9. Doft et at.9 en 1987 reportaron los resultados de 36 vitrectomías realizadas en 30 ojos de 17 pacientes con amiloidosis vítrea. La recurrencia de opacidades fue la causa más frecuente de reintervención y se presentó en el 27% de los pacientes. Las complicaciones de la vitrectomía incluyeron desprendimiento de retina que requirió manejo con cerclaje escleral en el 17% de los ojos y glaucoma que requirió cirugía filtrante en el 17% de los ojos.
Después de un seguimiento post operatorio medio de 35 meses, el 48% de los ojos tenían agudeza visual de 20/40 o mejor y el 32% de los ojos tenían agudeza visual entre 20/50 y 20/100. El 20% de los ojos tenían agudeza visual de 20/200 o peor debido a desprendimientonde retina, glaucoma de ángulo abierto u opacificación residual. Con el desarrollo de nuevas tecnologías y con el uso de técnicas menos invasivas, se ha reducido de manera importante la incidencia de complicaciones intra y post operatorias.
![]()
La vitrectomía 27 Gauge, ha demostrado ser una técnica segura en aquellos casos en los que hay una pérdida importante de visión o cuando hay una opacidad de medios queno permite hacer una adecuada valoración del fondo de ojo y que mejora significativamente los síntomas referidos por el paciente y la agudeza visual en la mayoría de los casos. Adicionalmente, se considera el tratamiento de elección en pacientes con glaucoma que ya han sido sometidos a trabeculectomia10.
Las principales dificultades en el manejo quirúrgico actual consisten en la remoción incompleta del vítreo y la recurrencia postoperatoria de las opacidades vítreas que se genera por una dispersión de las fibras residuales y por la continua producción de transtirretina en la retina y el plexo coroideo. Existe alto riesgo de rupturas retinianas iatrogénicas debido a las fuertes adherencias vitreorretinianas especialmente en el área de los vasos. Se debe hacer una inspección minuciosa de la retina periférica antes de terminar el procedimiento y en caso de encontrar una ruptura iatrogénica tratarla con fotocoagulación o criopexia dado el alto riesgo de desprendimiento de retina. Los depósitos de amiloide encontrados en los tejidos son porciones de inmunoglobulinas de cadena ligera.
Sin embargo, macroscópicamente pueden ser difíciles de diferenciar de material vítreo filamentoso normal. Las paredes de los vasos sanguíneos de los vasos sanguíneos coroideos y retinianos pueden estar engrosadas también por estos depósitos. Histológicamente los depósitos de amiloide en el vítreo se aprecian como material eosinófilo pálido, mostrando afinidad por el azul de toluidina y el cristal violeta (magenta oscuro). Con la tinción de Rojo Congo, el amiloide adopta una tonalidad naranja oscuro, mostrando birrefringencia y dicroismo al ser observado bajo luz polarizada11.
Reportamos el caso de una paciente con amiloidosis familiar diagnosticada a través de inmunotinción en muestra de vítreo cuya manifestación inicial y única fue la presencia de opacidades vítreas con estudios sistémicos negativos. Debido a que las manifestaciones oculares de la amiloidosis suelen aparecer años después del inicio de la enfermedad, el vítreo no ha sido considerado como material de primera opción en biopsias con fines diagnósticos. Sin embargo, el procesamiento y tinción de estas muestras debe considerarse en pacientes con presentación temprana de opacidades vítreas, resultados negativos de biopsia sistémica o curso atípico de la enfermedad.
Doctores:
Ana Mercedes García Albisua, alta especialidad en córnea y cirugía refractiva APEC
Renata García Franco, Marlon García Roa, Alejandro Arias Gómez, Sonia Corredor
Casas, Ximena Mira Lorenzo y Miguel Vázquez Membrillo
Instituto Mexicano de Oftalmología (Querétaro)
Bibliografía
Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-54.
Lachmann HJ, Hawkins PN. Systemic amyloidosis. Curr Opin Pharmacol. 2006;6(2):214-20.
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21(4):221-4.
Shukla S, Cohen A, Josephberg RG. Nonfamilial vitreous amyloidosis diagnosed by portable sutureless vitrectomy. Retin Cases Brief Rep. 2008;2(4):264-5.
Beirão JM, Malheiro J, Lemos C, Beirão I, Costa P, Torres P. Ophthalmological manifestations in hereditary transthyretin (ATTR V30M) carriers: a review of 513 cases. Amyloid. 2015;22(2):117-22.
Venkatesh P, Selvan H, Singh SB, Gupta D, Kashyap S, Temkar S, et al. Vitreous Amyloidosis: Ocular, Systemic, and Genetic Insights. Ophthalmology. 2017.
You J. Vitrectomy for vitreous amyloidosis. Int J Ophthalmol. 2011;4(3):307-10.
Hashemian H, Jabbarvand M, Khodaparast M, Khalilipour E, Esfehani HR. Ocular Presentations of Amyloidosis: InTech; 2013 [Available from: https://www.intechopen.com/books/amyloidosis/ocular-presentations-of-amyloidosis.
Doft BH, Machemer R, Skinner M, Buettner H, Clarkson J, Crock J, et al. Pars plana vitrectomy for vitreous amyloidosis. Ophthalmology. 1987;94(6):607-11.
Miyahara T, Ohta K, Yamamoto Y, Ueno A, Murata T. 25-gauge vitrectomy to treat ocular complications of familial amyloid polyneuropathy. J Glaucoma. 2007;16(1):169-70.
Latasiewicz M, Adan A, Solé M. Immunostaining images of vitreous transthyretin amyloid. Can J Ophthalmol. 2015;50(5):384-7.